Overview
B-Star is a web server for high-resolution HLA class I typing from Oxford Nanopore whole-gene amplicon sequencing data. The server accepts compressed FASTQ files and performs an end-to-end workflow including read preprocessing, alignment, phasing, allele-specific consensus reconstruction, HLA allele assignment, quality-control assessment, and report generation. Output reports include final HLA calls, supporting QC metrics, pharmacogenomic annotations, and embedded visual inspection with IGV.
B-Star is free and open to all users through the public web interface, and no login is required.
Pipeline overview
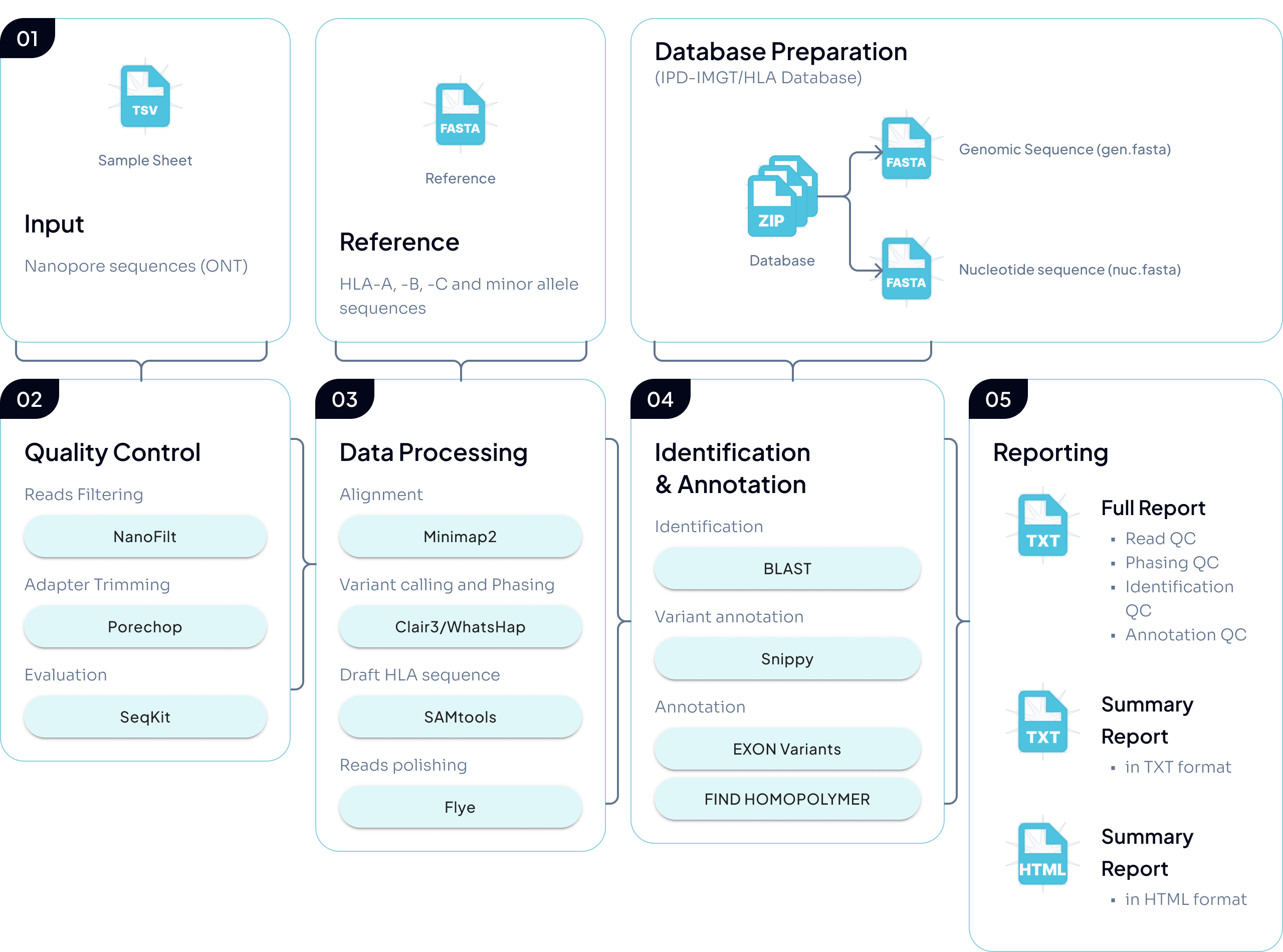
The current B-Star workflow consists of five main stages:
1. Input
B-Star takes ONT nanopore reads (in .fastq.gz or .fq.gz) as input.
2. Quality control
Reads undergo filtering/trimming and QC checks using standard tools (e.g., SeqKit and Porechop).
3. Alignment, phasing, and consensus reconstruction
Reads are first aligned to HLA reference sequences and variants are called with Clair3. Heterozygous variants are then used for allele phasing with WhatsHap, which partitions reads by allele. Finally, each partition is used to reconstruct an allele-specific consensus sequence with Flye-polish (with supporting processing/formatting via tools such as SAMtools).
4. Allele identification and variant annotation
For each phased allele, the consensus sequence is searched against the IPD-IMGT/HLA reference database using BLASTN to identify the best-matching reference allele. Differences between the consensus and the matched reference are then evaluated using SnpEff.
- If there are no mismatches, or mismatches occur only in intronic regions, the allele annotation is reported as high quality.
- If a few mismatches are detected in exonic regions, the annotation is flagged (Inspect) and we recommend manual inspection of the allele assignment and supporting evidence.
5. Report generation and result review
B-Star generates user-friendly output reports in both HTML and TXT formats. Summary reports are provided as summary_report_final.html and summary_report_final.txt, containing the final HLA genotype calls together with key quality-control metrics for result assessment. A more detailed QC report is also generated in TXT format as summary_fullreport.txt.
Software and database components
Third-party software
| Software | Role | Version | Licence | GitHub |
|---|---|---|---|---|
| SeqKit | Read filtering and summary statistics | v2.9.0 | MIT License | https://github.com/shenwei356/seqkit |
| Porechop | Adapter trimming | v0.2.4 | GPL-3.0 | https://github.com/rrwick/Porechop |
| Minimap2 | Long-read alignment | v2.28-r1209 | MIT License | https://github.com/lh3/minimap2 |
| Clair3 | Variant calling | v1.0.0 | BSD-3-Clause | https://github.com/HKU-BAL/Clair3 |
| WhatsHap | Read phasing and haplotype partitioning | v2.2 | MIT License | https://github.com/whatshap/whatshap |
| SAMtools | BAM processing | v1.13 | MIT/Expat | https://github.com/samtools/samtools |
| Flye | Consensus reconstruction | v2.9.2-b1786 | BSD-3-Clause | https://github.com/mikolmogorov/Flye |
| BLAST | Allele sequence matching | v2.12.0 | Public-domain software distributed by NCBI | https://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/2.12.0/ |
| SnpEff | Functional annotation of mismatches and gaps | v5.2 | MIT License | https://github.com/pcingola/SnpEff |
| IGV | Embedded interactive visualisation of alignments, variants and gene structure | V3.7.0 | MIT License | https://github.com/igvteam/igv.js |
Reference databases and annotation resources
- IPD-IMGT/HLA Database
Role: Reference HLA database used for read mapping, allele identification, and annotation database construction in B-Star (release v3.59.0).
Term usage: Used as an external reference resource for analysis and allele assignment. Copyrightable parts are distributed under CC BY-ND; attribution is required, and redistribution of modified versions requires permission.
Reference:
- Barker D, Maccari G, Georgiou X, Cooper M, Flicek P, Robinson J, Marsh SGE The IPD-IMGT/HLA Database Nucleic Acids Research(2023), 51(D1): D948-D955
- Robinson J, Barker D, Marsh SGE 25 years of the IPD-IMGT/HLA Database. HLA(2024),103(6): e15549
- Robinson J, Malik A, Parham P, Bodmer JG, Marsh SGE: IMGT/HLA - a sequence database for the human major histocompatibility complex Tissue Antigens (2000), 55:280-287
- PharmGKB / ClinPGx
Role: External pharmacogenomic annotation resource used to report drug-related information associated with inferred HLA alleles.
Terms of use: Used as an external annotation resource in the B-Star report. ClinPGx/PharmGKB data are distributed under CC BY-SA 4.0.
Licence and usage notice
B-Star is freely accessible through the public web interface. The server integrates third-party software and reference resources that remain subject to their respective licences and terms of use.
Output interpretation
B-Star reports inferred HLA haplotypes together with QC metrics used to assess typing reliability, including total read count, allele-specific read depth, percent identity, and counts of mismatches and gaps that may affect the encoded protein sequence. QC status is displayed as Pass, Inspect or Fail. The report also includes pharmacogenomic annotations and an embedded IGV view for manual review of alignments, variants and mismatch positions.
Example 1. Running the test_data.fastq.gz example
Users can evaluate B-Star by clicking "Try out sample file" at the bottom on the upload page, which automatically loads an example FASTQ file (test_data.fastq.gz) and sample sheet into the web server. After submission, the analysis proceeds through parameter selection and review, and the server returns an HLA typing report with QC metrics and drug hypersensitivity annotations. In this example, the result shows HLA-B*15:02:01 and HLA-B*58:01:01, and all QC metrics are marked as Pass. This indicates that the allele assignments are well supported by the sequencing data and can be interpreted as reliable. The report also provides direct links to the IGV view and consensus sequence files for further inspection.

Example 2. Inspecting a borderline HLA call using IGV
This example uses ERR11404057.fastq.gz from the SRA, corresponding to NA17280 in our manuscript. In the R9.4.1 / Matern / Rapid dataset, B-Star reports a homozygous HLA-B*08:01:01 result, but one allele is marked as Inspect because of multiple mismatches. By opening the IGV view, users can see that many mismatches cluster near the beginning of the allele, consistent with non-specific mapping, and can also review highlighted coding variants such as missense or frameshift changes. This result suggests that stricter filtering or a more specific primer set may be needed.
